



PAMサイトの検索とgRNAの設計
CRISPR(Clusters of Regularly Spaced Interspersed Short Palindromic Repeats)-Cas9システムは、細菌においてウイルスなど遺伝的要素の侵入物を特異的に排除するよう進化した獲得免疫機構です。この機構を利用して、二本鎖DNAを切断してゲノム配列の任意の場所を削除・置換・挿入することができるようになりました。
大きな配列からgRNAの候補を検索し、それらをスコア化するにはオンラインのgRNAデザインツールを使用する必要があります。しかし、比較的小さな配列でゲノムターゲットが限定されている場合は、SnapGeneを使用してgRNAを設計することができます。
このチュートリアルでは、SnapGeneでPAMサイトの検索とgRNAの設計を行う方法をご紹介します。
1.【準備】DNA配列を入手する
任意の配列(500~700 bpぐらいの配列)を各自用意してください。本チュートリアルでは以下の配列を使用します。
SnapGeneにDNA配列をコピーします。SnapGeneの起動から配列のコピーまでは、SnapGeneラーニングの プラスミドマップ:プラスミドデータの取得とマップの表示 を参照してください。
1-1. フィーチャータイプを追加
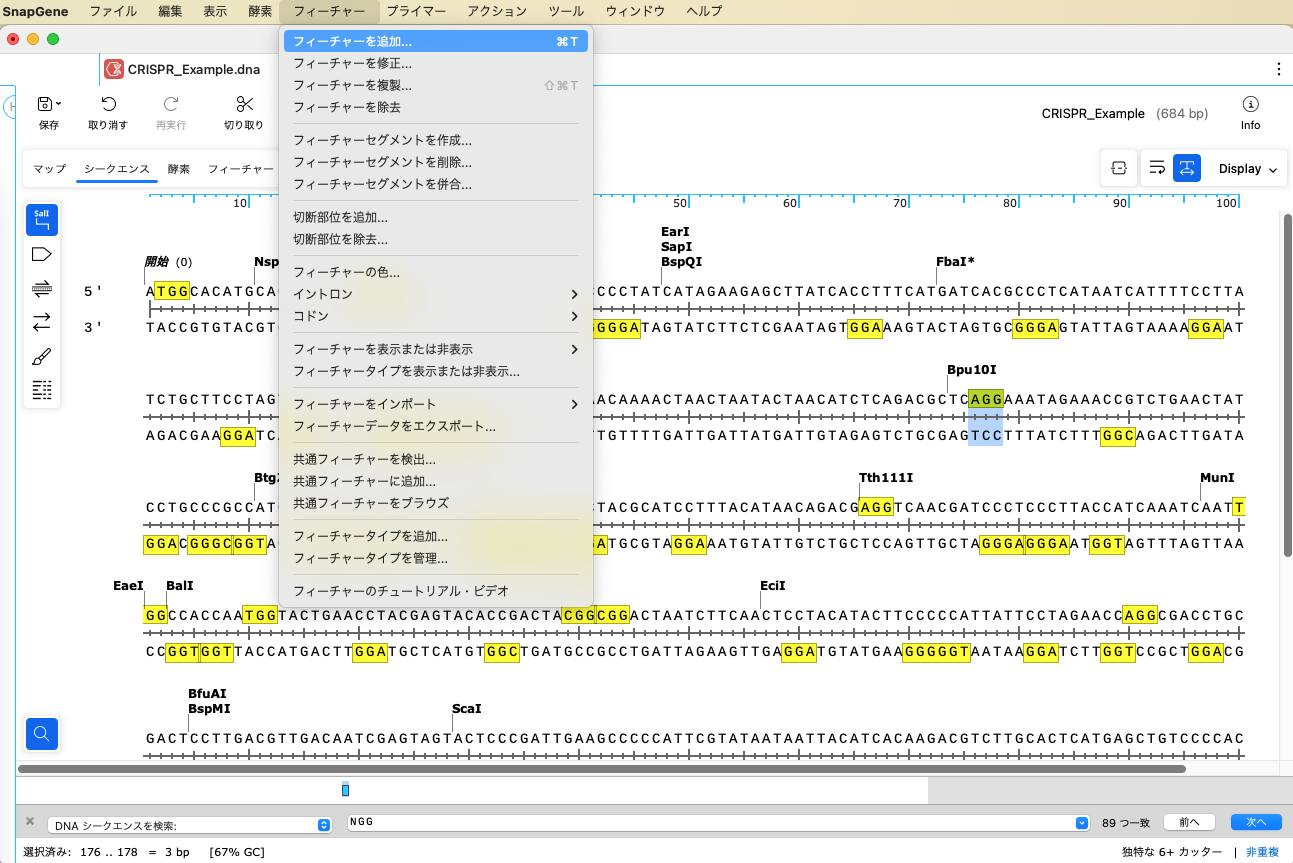
チュートリアルでは PAM配列にフィーチャーを追加します。カスタムフィーチャー機能を使用して、あらかじめフィーチャータイプを作成しておきます。「フィーチャー」メニューから、「フィーチャータイプを追加」を選択します。
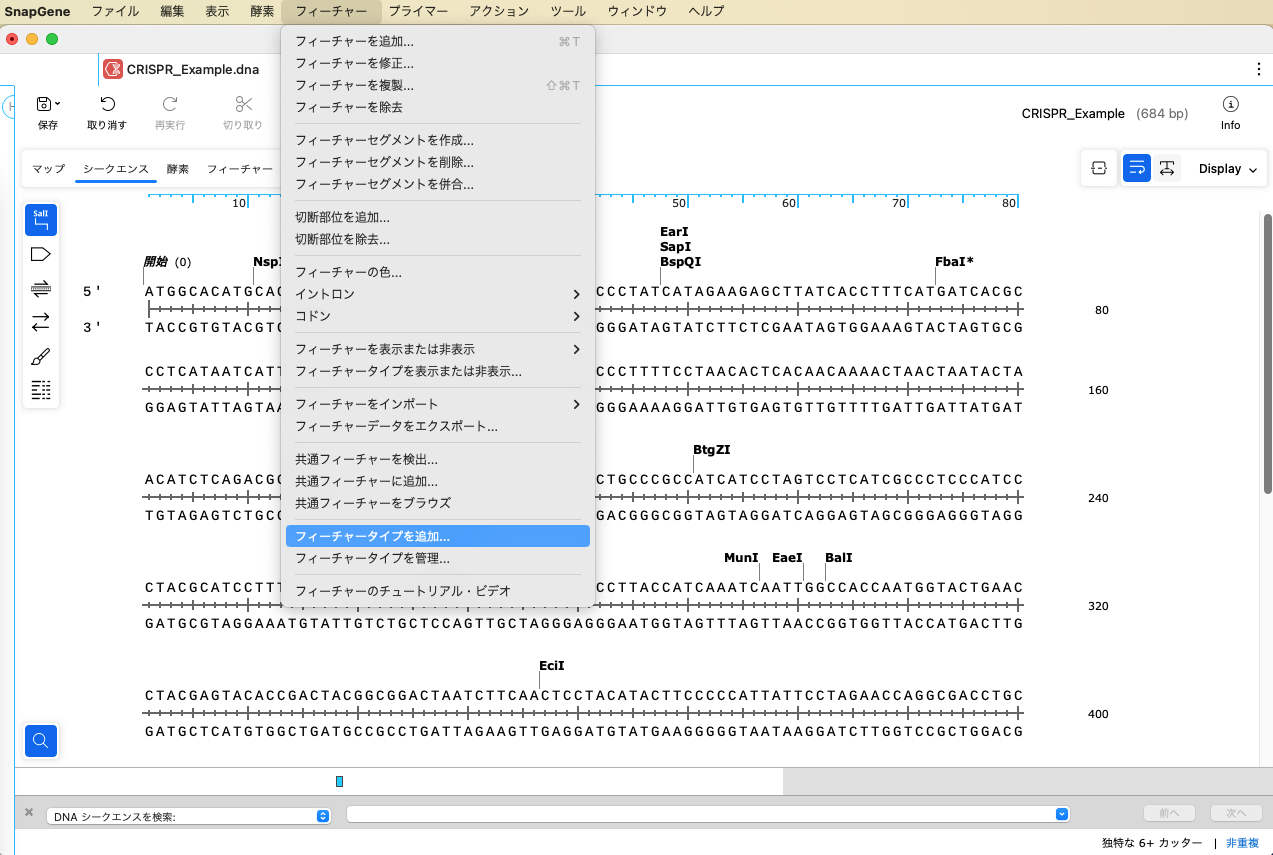
「フィーチャータイプを追加」ダイアログで、「タイプ」1 にタイプ名を入力します(ここでは、"CRISPR_gRNA")。「デフォルトの色」2 で任意の色を選択し、OKボタンをクリックします。これで、フィーチャータイプに CRISR_gRNA が追加されました。
2. PAMサイトの検索とgRNAの設計
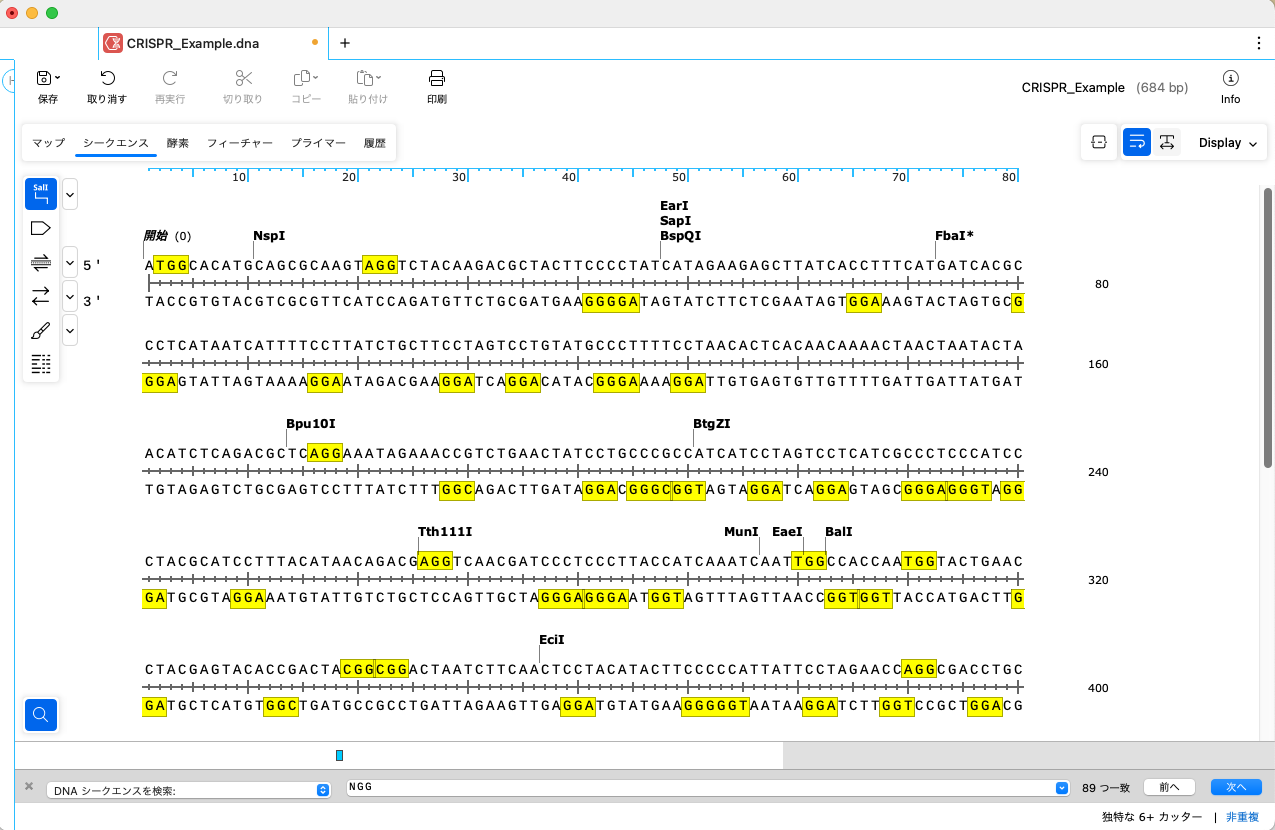
シークエンスタブを開き、メニューバーの「編集」→「検索」→「DNAシークエンスを検索」を選択します。
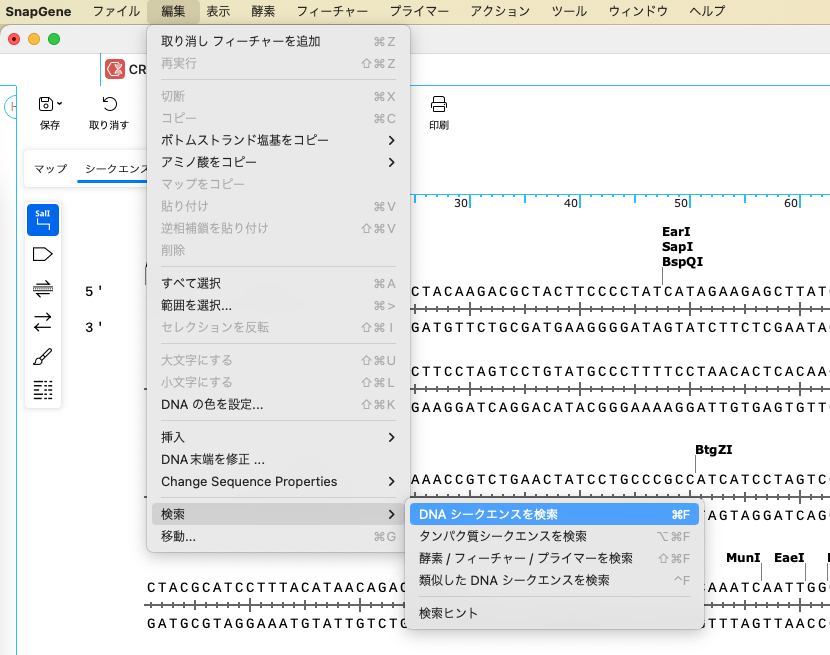
ウィンドウの下部に検索フィールドが表示されるので、"NGG" と入力しPAMサイトを検索します。検索フィールドは、Control + F のショートカットキーでも表示することが可能です。
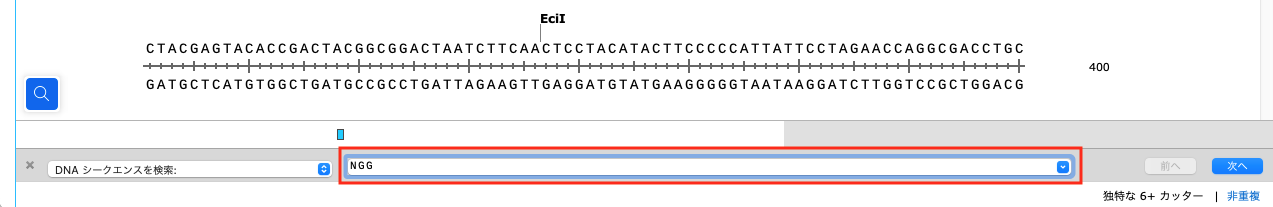
両鎖のすべてのPAMサイトがハイライトされます。なお、NGGは 5′-NGG-3’ の規則に一致するように定義されています。
フィーチャー機能を使って、実験に適した NGG を PAM Site と表示しましょう。
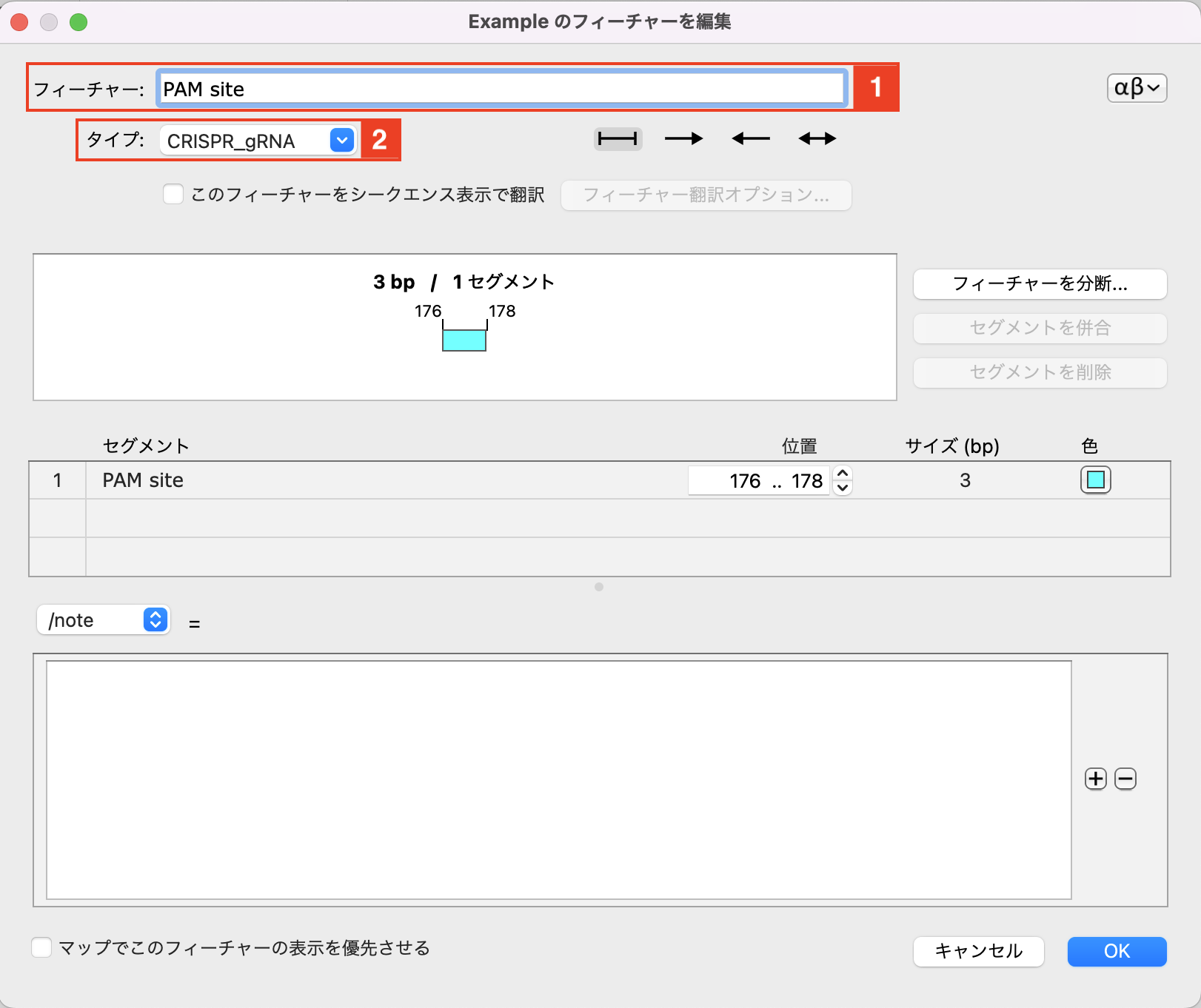
ここでは、176〜178塩基目の AGG を選択します。176〜178塩基を選択後、メニューバーの「フィーチャー」→「フィーチャーを追加」を選択します。
フィーチャー名は "PAM Site" にします。フィーチャータイプで "CRISPR_gRNA" を選択し、OKボタンをクリックします。
シークエンスにフィーチャー(PAM Site)が追加されました。
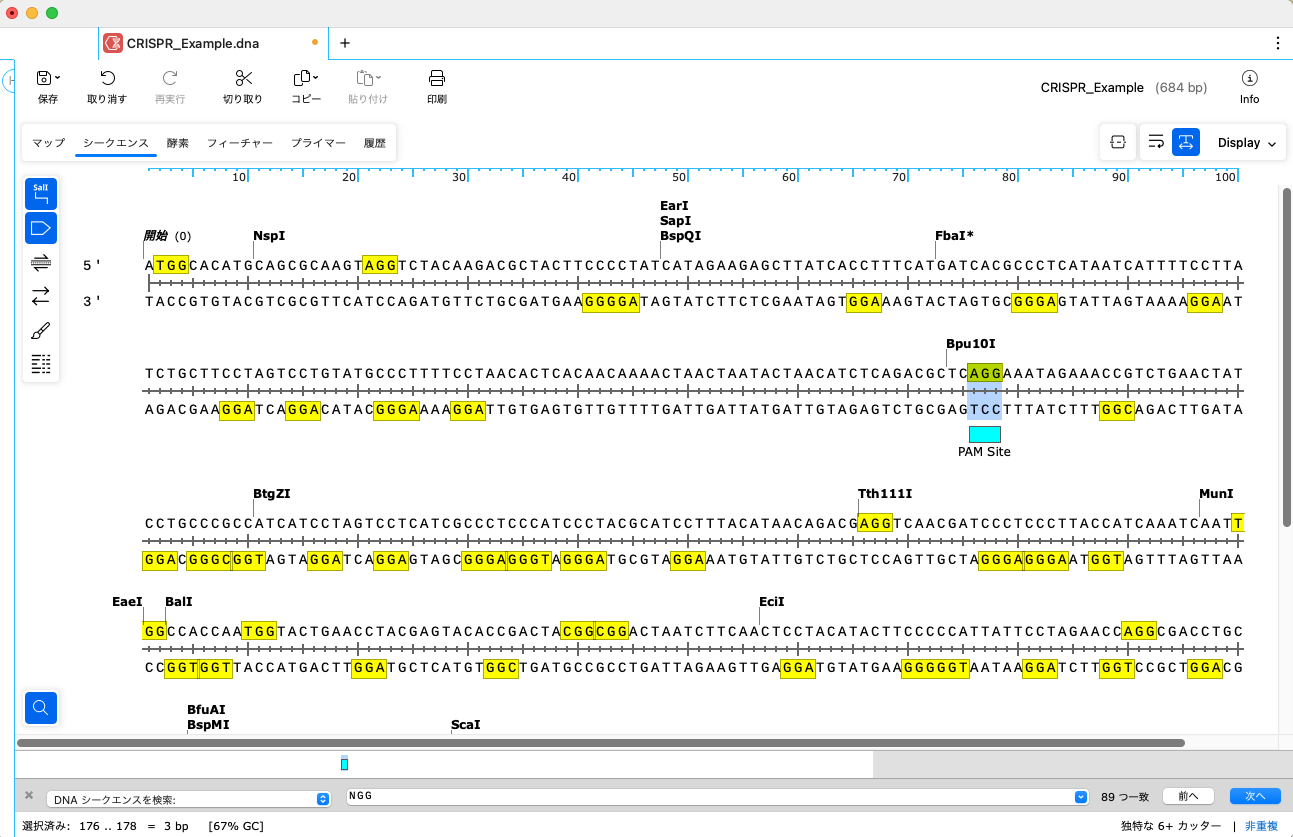
PAMサイトの 5’側にある 20個のヌクレオチドを特定します。この 20塩基がホーミング装置として働き、PAM site の直前に位置する特異的DNA標的部位へ Cas9/gRNA 複合体を動員します。

SnapGene プライマー機能やフィーチャー機能を使用して、これらの配列にラベルを付けます。
メニューバーの「プライマー」から「プライマーを追加」をクリックすると、任意のプライマーを追加することができます。

PAM配列から 3塩基内側にあるターゲット配列が Cas9 切断部位です。必要に応じて、切断部位を登録します。

以上が SnapGene を利用して行える機能になります。
CRISPR-Cas9 を使用する際に気にすべき事項として、オフターゲット作用があります。オフターゲット作用は、本来期待していない部位に挿入・欠損・変異が生じ、得られた表現型が損なわれてしまう現象です。
残念ながら、SnapGene では CRISPR-Cas9 システムの特異性を検証することはできません。ただ、CRISPR-Cas9 システムのオフターゲット作用の頻度を予測することは困難です。また、対象配列の領域が決定している場合は、候補となるPAMサイトが限られており、CRISPR-Cas9 システムの効率を検証することもできません。以上より、比較的小さな配列でゲノムターゲットが限定されている場合は、今回のような方法で行うことが現実的であると考えられています。
3. 相同組換え修復テンプレート
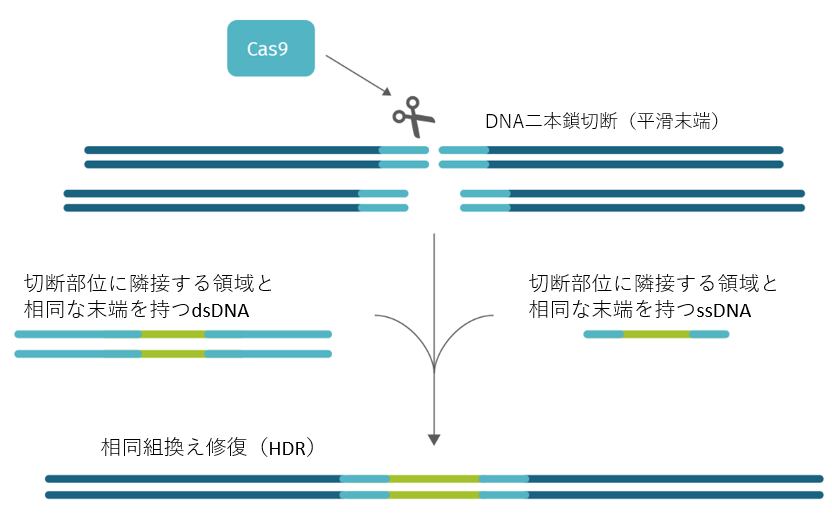
CRISPR/Cas9 を応用すると特定のゲノム遺伝子座に DNA 配列を置換・挿入するノックインすることもできます。これを相同組換え修復(homology-directed repair [HDR])による遺伝子ノックインと呼びます。この場合、Cas9 タンパク質と gRNA に加えてノックインドナーとなる DNA が必要となります。そして、ドナーテンプレートは、切断部位に隣接する領域と十分な相同性を持つ必要があります。
SnapGeneのタンパク質翻訳ツールを使うことで、HDR ドナーテンプレートの設計などゲノム編集がタンパク質発現に与える影響をシミュレーションすることができます。
HDR による遺伝子ノックインのシミュレーションは、大きく2つのステップで構成されています。
ステップ1.ゲノムの編集を定義する
SnapGene では DNA配列の編集を簡単に行うことができ、すでに DNAファイルに添付されているすべての注釈情報で編集内容を定義することが可能です。編集メニューやトップツールバーから、頻繁に使用する編集ツールに直接アクセスすることができます。
さらに、View Menu や Side Toolbar からアクセスできるタンパク質翻訳ツールを使って、ゲノム編集がタンパク質発現に与える影響をテストすることもできます。
ステップ2.HDR テンプレートを入手する
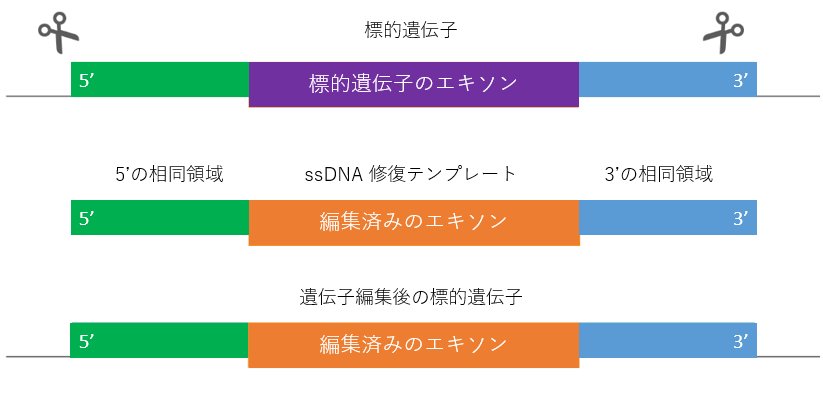
ノックインに使用する HDR テンプレートには、二本鎖DNA(dsDNA)と 一本鎖DNA(ssDNA) があります。ゲノム編集の位置とサイズに応じて、どちらのタイプの修復テンプレートを設計するか決定する必要があります。
ゲノム編集の長さが200ヌクレオチド未満の場合
ゲノム編集のサイズが小さい場合、ssDNA の形で HDR テンプレートを提供することができます。メーカーにより設計のガイドラインは異なりますが、通常 80 ~ 200 ヌクレオチドの範囲となります。
このサイズの修復テンプレートは、対応できる編集の種類に制限があります。しかし、dsDNA と比較して ①細胞毒性が低く、②オフターゲットが生じる可能性を抑えることが可能と報告されています。
PAM部位は ssDNA の中央に配置し、PAM の近くでヌクレオチドを変化させる必要があります。変異は、PAM部位を破壊する変異を含み、修復に成功した後、Cas9 切断の影響を受けなくなるようにする必要があります。
ゲノム編集の長さが 200 塩基以上の場合
ゲノム編集の長さが 200 塩基以上の場合、2つの方法があります。
クローニングによるHDRテンプレートの構築
遺伝子の大きな部分をノックインまたはノックアウトしたい場合、分子クローンとして修復テンプレートを設計し、場合によっては構築する必要があります。この方法でコンストラクトの構築するには、遺伝子クローニング技術を用いることが一般的です。
HDR テンプレートに 800 bp までの長さの相同性アームを含める必要があり、またアームにはリピート配列を含めないようにしてください。
SnapGene の Actions メニューにあるクローニングシミュレーションツールを使用すると、分子試薬との関連においてクローニング戦略の結果を適切に設計および予測することができます。
合成 DNA 分子の発注によるHDRテンプレートの構築
長い一本鎖DNA(ssDNA、lssDNA、メガマー)を合成し、配列を確認することができ、その長さは現在 2000 ヌクレオチドまで伸びています。相同性アームは 100~400 ヌクレオチドが推奨されています。
100 ヌクレオチドの相同性アームを含む1000 bp の lssDNA を用いた効率的なゲノム編集も報告されています。このゲノム編集の成功の鍵の一つは、修復テンプレートの両端を二本鎖切断にすることです。
以上で、PAM サイトの検索とgRNA の設計を終わります。